To become or remain competitive, companies must constantly innovate by developing new products. Unfortunately, many development projects fail prematurely, it is important to use a systematic and strategic product development process, to reduce ineffective deployment of resources.
Product development projects have many types of risks. In terms of development time and costs, in terms of technical feasibility, competitive products, obtaining market access (being allowed to place the product on the market), legislations and regulations that the product must comply with and not having the right knowledge and skills in-house.
To limit the risks, it is advisable to phase the development project according to a product development model such as the Phase Gate Process model. Each phase has its own mandatory documents, and each phase has its own go/no go moments.

This Q&A describes high level basic requirements, tips and pitfalls, companies encounter during the development and distribution of a medical device. The operation, advantages, and disadvantages are only explained to a limited extent, including why and how having a strategic development model strengthens the company’s quality management system (QMS).
What is needed for the technical documentation?
In accordance with Article 10.4 MDR (EU) 2017-745, the manufacturer of a medical device is obliged to draw up a technical file (TD) and keep it up to date. The content of the TD is described in Annex II and III of the MDR (EU) 2017-745.
Having a fixed format on a network drive, where all the necessary technical documentation can be stored, provides an overview, and helps collect and categorize the information for the Notified Body. Please note that each development phase should have its own folder structure. This ensures that it is easy to find out in which development phase what happened. Note, creating files from scratch, linking requirements and risks, and keeping track of all changes is not an easy task. It is smart to use a ‘medical device document management system’ to keep track of the required documentation. There are a lot of suppliers that provide this kind of software, you should create a list of requirements/ features that the document management software should offer.
Since the TD and the QMS must be assessed/audited by a Notified Body for most medical device software and it is unlikely that the Notified Body its auditors can speak the Dutch language, you need to draw the required documentation up in the English language.
During an QMS audit, or during product certification, the auditor may require evidence that the development plan has been followed or explain why certain development choices have been made. To prevent documents from being unusable, when they are written in Dutch, it is advisable to draw up all documents, including meeting minutes, in the English language. If there are operational reasons to have documents available in Dutch, such as assembly instructions, they you can deviate from this rule. However, the test protocols (such as IAT and FAT) will have to be available in English. These are quality records.
What are the design history file and design master records?
In order to get a medical device certified to be placed on the market, the MDR contains documents that the legal manufacturer must provide. To ensure that the TD is complete, and to monitor the progress of writing the TD documents, it is useful to make a checklist of the documents to be delivered, which can then be allocated to a specialist per part. When the checklist is expanded with start and expected end dates, this list can be used as an action list to monitor the progress of documentation.
The TD must be divided into design choices made during the development of prototypes (the design history file) and the technical data of the end product (design master record). Having a Phase Gate Process model makes the control and construction sequence of these files a lot easier. When it has been demonstrated that the product idea is both economically and technically feasible, which has been demonstrated in phases 0 and 1, the development documents that are generated in phases 2 and 3 are the documents that will be placed in the design history file.
At the end of each product development phase, an evaluation should be made about the progress of the project. The findings are described and report, after sharing with the steering committee, is added to the project file. The lessons learned should be addressed in the QMS (as part of continuous improvement).
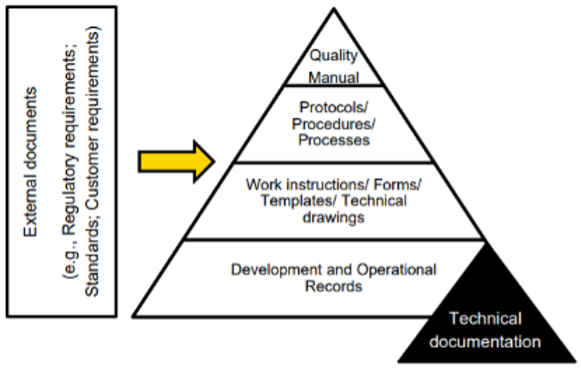
At the end of phase 3 is the ‘Design Transfer’ takes place[1]. The product is production ready meaning that all technical drawings are available, the bill of materials has been drawn up, the safety critical components have been identified and the device has been verified and validated[2]. The documents required for the production of the first production series are placed in the design master record. Note, all technical documents that are drawn up from this point are placed in the design master record. The contents of the design history file and design master record, which will be described later.
Having a phase-out model to stop technical support of the device is an important part of the quality management process, especially since mandatory post-market surveillance can be organized through this model (timelines, frequency, and reporting (periodic safety update reporting (PSUR)).
The safety critical components, as listed in the TD, (provided and approved by the Notified Body), require periodic checks, to determine if the datasheets are still the latest versions, and the approval marks are still valid. Note, it is not allowed to change the safety critical components without the approval of the Notified Body.
What is needed for a quality management system?
The development of a medical device must follow a demonstrably assured process. Having a Quality Management System (QMS) in place is mandatory and is audited by the Notified Body. The legal manufacturer of a medical device draws up, for each product that will be developed and for each market that product will be placed on the market, a quality- and regulatory plan. In this plan the quality management methodology must be described. The choice must be made whether the QMS will be certified according to the international ISO13485:2015 standard, or whether it will be chosen not to have the QMS certified, but to have the Notified Body assess the QMS during the product certification. Do make sure that the QMS has a document-based structure, since you will need to be able to demonstrate the interaction between the processes and on what basis your QMS is founded. Quality records will provide evidence that the quality policy is followed.

Do I need QMS certification?
QMS certification has the advantage that only one audit needs to be carried out for the CE certification of the medical device. If the manufacturer of the medical device wants to bring multiple variants or different products to the market, ISO13485 certification is more (costs) efficient. If the product of a medical device is a one-off activity, the manufacturer can choose to set up the QMS in accordance with the ISO13485 standard, but not to have it certified. During the product certification process, two audits (stages 1 and 2) will be carried out by the Notified Body to assess the QMS. For an ISO13485 certified product, this will be one audit.
The biggest risk of not certifying is the time lost if it turns out that the QMS does not meet the requirements. Implementing an improvement plan (corrective action plan) to ensure a major non-conformity is effective, measurable, and verifiable can take several months. As a result, the marketing plan (phase 4 and 5) cannot be implemented.
[1] ISO13485:2016 (7.3.8)
[2] ISO13485:2016 (7.3.5, 7.3.6)
